
Mitochondrial Biochemistry, Genetics, and Medicine
Welcome to the Gohil Lab Homepage www.youtube.com/watch?v=CkHqfF0SatY&list=PLasOe0z6Zl9rd72QFaJajh8hkijGSwiIN. Our laboratory is located in the Interdisciplinary Life Science Building of the Texas A&M University. We are a part of the Department of Biochemistry and Biophysics as well as the Program in Genetics at Texas A&M University, College Station.
Our laboratory studies mitochondria, the cellular powerhouses that are present in almost all eukaryotes. These ancient organelles, besides producing ATP energy, play a central role in cell signaling, intermediary metabolism, ion homeostasis, and even in cell death. Aberrant mitochondrial energy metabolism has been associated with numerous age-related human diseases including cancer, diabetes, and neurodegenerative disorders, as well as monogenic metabolic disorders, which typically manifest in newborns and young children. In fact, about 20% of predicted human mitochondrial proteins have been implicated in one or more inherited diseases.
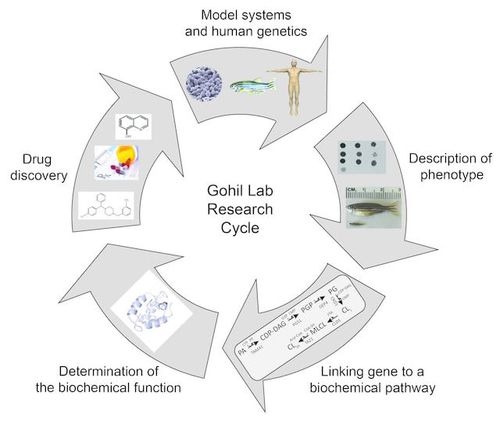
Despite the fundamental role of the mitochondrion in cellular energy metabolism and its involvement in numerous human diseases, we still do not know the full list of proteins or the lipids required for building the mitochondrial respiratory chain (MRC) – an ancient biochemical pathway necessary for energy production. We use an integrative approach based on clues from evolutionary history, subcellular proteomics, and human genetics to discover novel mitochondrial proteins required for MRC biogenesis. Our approach is based on the simple premise that since core components of the MRC are conserved, the assembly factors required to build the respiratory chain should also be conserved. Therefore, we utilize multiple model systems, including yeast, zebrafish, and human patient cell lines, to determine the role of these conserved proteins and phospholipids in mitochondrial bioenergetics, organismal development, and human disease pathogenesis, respectively.
With this approach, we have discovered a family of proteins required for copper delivery to one of the MRC complexes and determined the specific phospholipid requirements for MRC function and formation. Using our genetic models of mitochondrial disorders of copper and phospholipid metabolism, we aim to discover pharmacological agents and endogenous metabolites that may emerge as novel therapeutics for mitochondrial disorders by restoring MRC function in these models.
Our laboratory studies mitochondria, the cellular powerhouses that are present in almost all eukaryotes. These ancient organelles, besides producing ATP energy, play a central role in cell signaling, intermediary metabolism, ion homeostasis, and even in cell death. Aberrant mitochondrial energy metabolism has been associated with numerous age-related human diseases including cancer, diabetes, and neurodegenerative disorders, as well as monogenic metabolic disorders, which typically manifest in newborns and young children. In fact, about 20% of predicted human mitochondrial proteins have been implicated in one or more inherited diseases.
Despite the fundamental role of the mitochondrion in cellular energy metabolism and its involvement in numerous human diseases, we still do not know the full list of proteins or the lipids required for building the mitochondrial respiratory chain (MRC) – an ancient biochemical pathway necessary for energy production. We use an integrative approach based on clues from evolutionary history, subcellular proteomics, and human genetics to discover novel mitochondrial proteins required for MRC biogenesis. Our approach is based on the simple premise that since core components of the MRC are conserved, the assembly factors required to build the respiratory chain should also be conserved. Therefore, we utilize multiple model systems, including yeast, zebrafish, and human patient cell lines, to determine the role of these conserved proteins and phospholipids in mitochondrial bioenergetics, organismal development, and human disease pathogenesis, respectively.
With this approach, we have discovered a family of proteins required for copper delivery to one of the MRC complexes and determined the specific phospholipid requirements for MRC function and formation. Using our genetic models of mitochondrial disorders of copper and phospholipid metabolism, we aim to discover pharmacological agents and endogenous metabolites that may emerge as novel therapeutics for mitochondrial disorders by restoring MRC function in these models.
Yeast Cells
|

Fish
|

Mice
|
Human Cells
|
Our team consists of research scientist, postdoctoral researchers, graduate students as well as undergraduates. We strongly believe in collaborative science, with emphasis on training and mentoring. The laboratory is currently supported by funds from the National Institutes of Health, American Heart Association and the Welch Foundation.